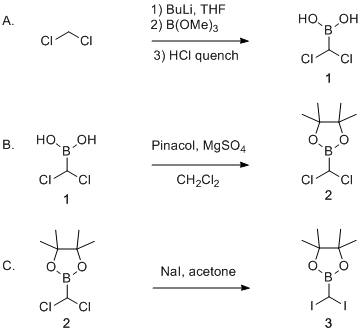
A. (Dichloromethyl)boronic acid (1). A flame-dried 1-L, three-necked, round-bottomed flask (Note 2) is equipped with a 125-mL addition funnel fitted with an argon inlet, an overhead mechanical stirrer with a 20 x 75 mm Teflon paddle (Notes 3 and 4) and a rubber septum fitted with an argon inlet, an argon outlet (Note 5) and a temperature probe (Note 6). The flask is charged by cannulation of dry tetrahydrofuran (200 mL) (Notes 7 and 8) and by syringe with dry dichloromethane (10.0 mL, 156 mmol, 1.23 equiv) (Note 9) (Figure 1).
The reaction mixture is cooled to -100 °C in an ethanol/liquid nitrogen bath (Notes 10, 11 and 12) and stirred vigorously (400 rpm). A solution of n-butyllithium (2.5 M in hexanes, 50.7 mL, 127 mmol, 1.00 equiv) (Note 13) is charged in the addition funnel and is then added dropwise over 45 min while maintaining the internal reaction temperature at -100 °C (Figure 2) (Note 14). The reaction mixture is stirred at this temperature for 40 min (Note 15).
Trimethyl borate (15.0 mL, 133 mmol, 1.05 equiv) (Note 16) is added in one portion by syringe via the rubber septum equipped neck (Note 17). The reaction mixture is stirred at -100 °C for 40 min. Hydrochloric acid (5 N, 30 mL) is added in one portion at -100 °C, then the cooling bath is removed and the reaction mixture is stirred for 1 h (Notes 18 and 19). A cloudy white solution is obtained and it quickly becomes clear and colorless (Figure 3).
The solution is poured into a 1-L separatory funnel. The three-necked flask is rinsed with diethyl ether (2 x 50 mL), and the solution is transferred into the separatory funnel (Figure 4 (a)). After thorough mixing, the aqueous layer is separated and extracted with diethyl ether (2 x 50 mL). The combined organic layers are washed with brine (100 mL) and dried over 15 g of magnesium sulfate. The resulting homogeneous solution is filtered through an 11-cm diameter Büchner funnel (Figure 4 (b)) (Note 20). The colorless solution is transferred into a 1-L round-bottomed flask and concentrated under reduced pressure (320 mmHg, 25 °C) to remove diethyl ether then (130 mmHg, 25 °C) to remove tetrahydrofuran (Note 21). When most of the solvents are evaporated, the solution is transferred (using diethyl ether to rinse the flask) into a tared 250 mL round-bottomed flask and concentrated under reduced pressure (320 mmHg, 25 °C then 130 mmHg, 25 °C).
The crude is dried under reduced pressure on a high-vacuum pump (0.2 mmHg) for 1 h to afford desired boronic acid 1 (20.9 g, 78% w. purity (being 16.3 g, 127 mmol, quantitative yield) as a yellow-brownish syrup (Note 22) which is used with no further purification for next step (Notes 23, 24 and 25) (Figure 5).
B. 2-(Dichloromethyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (2). In the 250-mL round-bottomed flask containing crude (dichloromethyl)boronic acid 1 obtained from Step A (16.3 g, 127 mmol, 1 equiv) under argon is added a 5 cm egg-shaped magnetic stir bar and dry dichloromethane (100 mL) (Figure 6) (Note 26). After complete dissolution of the boronic acid, magnesium sulfate anhydrous (15.3 g, 127 mmol, 1 equiv) (Note 27) and pinacol (15.73 g, 133 mmol, 1.05 equiv) (Note 28) are added. The obtained heterogeneous reaction mixture is stirred under argon at room temperature for 16 h.
The reaction mixture is filtered through a 10.5 cm diameter sintered glass funnel over a pad of Celite (2 cm high) (Figure 7 (a)) (Note 29). The cake is washed with dichloromethane (2 x 50 mL). The filtrate is transferred into a 500 mL round-bottomed flask and concentrated under reduced pressure (270 mmHg, 25 °C). The obtained beige liquid is dried under reduced pressure on a high-vacuum pump (0.2 mmHg) for 1 h to remove any residual solvent (Figure 7 (b)).
The residue is transferred into a 40-mL Claisen flask equipped with a glass stopper, a glass thermometer fitted with a glass adapter and a 16-cm condenser on which is placed a distillation receiver with a 10-mL round-bottomed flask for the first fraction, a tared 50-mL round-bottomed flask for the second fraction and a tared 25-mL round-bottomed flask for the last fraction. Granules for smooth boiling (1.3 g) are added (Note 30). The product is distilled under reduced pressure through a 10-cm vacuum-jacketed distilling column (Figure 8) (Notes 31 and 32).
A forerun (ca 0.5 mL) is collected and discarded, and then desired boronic ester 2 is obtained (24.2 g, 89%) as a colorless liquid, distilling at 108 °C (23 mmHg) (Figure 9) (Notes 33, 34 and 35). The liquid turns into an amorphous, low melting solid upon standing in the freezer overnight.
C. 2-(Diiodomethyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (3). A 1-L flame-dried round-bottomed flask, equipped with a 24-cm condenser and a 6.5-cm egg-shaped magnetic stir bar, is charged with 2-(dichloromethyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane 2 obtained from Step B (22.8 g, 108 mmol, 1 equiv), sodium iodide (37.3 g, 249 mmol, 2.3 equiv) (Note 36) and dry acetone (230 mL) (Figure 10) (Note 37).
The obtained yellow solution is heated under reflux conditions at 60 °C in a heating mantle under argon for 48 h (Note 38). A sheet of aluminum foil is placed around the round-bottomed flask to protect the reaction from light (Figure 11) (Note 39).
The reaction mixture is cooled down to room temperature and filtered through a 14-cm diameter sintered glass funnel over a pad of Celite (2 cm high) to remove NaCl salts formed during the reaction. The cake is washed with acetone (500 mL) until the orange color disappears from the pad (Figure 12).
The obtained resulting red solution is transferred into a 1-L round-bottomed flask covered with a sheet of aluminum foil and concentrated under reduced pressure (155 mmHg, 25 °C) to give a grey-greenish solid (Figure 13).
Dichloromethane (300 mL) is added, giving an orange solution which is transferred into a 1-L Erlenmeyer flask fitted with a 6.5-cm octagon magnetic stir bar. Sodium thiosulfate pentahydrate (40 g) (Note 40) and anhydrous magnesium sulfate (45 g) (Note 41) are added in alternating portions of 5 g and 5.5 g, respectively, to quench residual iodide, until noting disappearance of the orange color (Figure 14).
The obtained pale yellow solution is filtered through a 14-cm diameter sintered glass funnel over a pad of Celite (2 cm high) (Note 42), transferred into a 1-L round-bottomed flask and concentrated under reduced pressure (300 mmHg, 25 °C) (Note 43). The obtained pale yellow solid is dried under reduced pressure on a high-vacuum pump (0.2 mmHg) for 2 h to afford desired boronic ester 3 (40.5 g, 95%) as a pale yellow solid (Note 44). The product can be used with no further purification or can be recrystallized.
Recrystallization is performed on the aforementioned batch. The solid is placed in a tared 250-mL round-bottomed flask and dissolved of hot hexanes (35 mL) (70 °C) (Figure 15).
The yellow solution is allowed to cool to room temperature, then cooled to 0 °C in an ice bath for 30 min to allow for slow crystallization. The yellow supernatant is removed by Pasteur pipette and transferred into a tared 100-mL round-bottomed flask. The solid is transferred into a 10.5-cm diameter sintered glass funnel and rinsed with 50 mL of cold hexanes (0 °C) (Note 45). The resulting solid is dried overnight under reduced pressure on a high-vacuum pump (0.2 mmHg) to afford pure boronic ester 3 (34.1 g, 80%) as a pale yellow crystalline solid (Figure 16) (Notes 46, 47 and 48).
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/content/acs/en/about/governance/committees/chemicalsafety/hazard-assessment.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
n-butyllithium,
dichloromethane,
tetrahydrofuran,
trimethyl borate,
hydrochloric acid,
diethyl ether,
pinacol,
sodium iodide,
magnesium sulfate,
calcium sulfate and
diphenylacetic acid.
2. All glassware is flame-dried under vacuum and purged with argon prior to use. All reaction steps are performed under a partial positive argon gas atmosphere using an argon gas line connected to an external mineral oil bubbler.
3. A mechanical stirrer is employed as magnetic stirring through the dewar is not sufficiently efficient.
4. The mechanical stirrer used is a EUROSTAR power control-visc, IKA-WERKE.
5. An argon outlet is placed on the rubber septum to ensure that argon is flowing through the equipment set-up.
6. The temperature probe (A VWR Traceable Waterproof Type-K Thermometer, Cat. #89094-738, with a low temperature type-K probe, Cat. #23609-238) is placed inside the reaction mixture in order to carefully monitor the internal reaction temperature.
7. Tetrahydrofuran was purified by passage through a column of activated alumina on a filtration system.
8. A 250-mL flame dried round-bottomed flask is charged with dry tetrahydrofuran, which is then transferred by cannula into the 1-L three-necked reaction flask.
9. Dichloromethane was freshly distilled over CaH2 before use.
10. The rate of the argon flow should be sufficiently high when adding liquid nitrogen in the bath to prevent the oil in the bubbler from being sucked-in the line.
11. When reaching -100 °C, ethanol solidifies. A spatula is used to break the solid that had formed and to obtain a gel. Achieving a stable internal temperature of -100 °C may take from 45 min to 1 h.
12. To obtain an internal temperature of -100 °C, the ethanol/liquid nitrogen bath should be maintained near - 120 °C.
13. The
n-Butyllithium solution (2.5 M in hexanes) was purchased from Sigma Aldrich and titrated with
diphenylacetic acid, 99% (purchased from
Sigma Aldrich and used as received) before use.
14. Liquid nitrogen is added regularly to maintain the internal temperature at -100 °C.
15. Black residues may appear on the wall of the flask due to dichloromethane carbene formation/polymerization.
16. Trimethyl borate, 99% was purchased from Alfa-Aesar and used as received.
17. After addition of trimethyl borate in one portion the internal temperature rises to -70 °C. Liquid nitrogen is added to the dewar in portions, waiting several minutes between additions, until the internal reaction temperature has returned to a stable -100 °C.
18. The reaction mixture is stirred for 1 h starting from the time when the ethanol/liquid nitrogen bath is removed.
19. The reaction mixture is placed in a room temperature water bath that is changed regularly to help the internal reaction temperature rise faster to room temperature.
20. A Fisher Q8 filter paper (Cat No. 09-790C, diameter: 9.0 cm) was used.
21. The colorless solution becomes pale yellow when concentrated.
22. The color of the crude mixture may range from yellow to reddish brown.
23. Significative amounts of tetrahydrofuran and diethyl ether remain in the product, but this residual solvent does not interfere in next step. The crude reaction mixture contained a mixture of 1, tetrahydrofuran and diethyl ether. The ratio of these compounds was determined by 1H NMR (400 MHz, CDCl3) by integration of the following diagnostic peaks: 1 (5.34 ppm, s, 1H), tetrahydrofuran (3.87 - 3.84 ppm, m, 4H) and diethyl ether (3.52 ppm, q, J = 7.0 Hz, 4H). A typical crude reaction mixture contained a 100:276:19 ratio of 1:tetrahydrofuran:diethyl ether.
24. A second reaction on the same scale afforded 21.0 g (78% w. purity) of 1 (quantitative yield).
25. The product has been characterized as follows: 1H NMR (400 MHz, CDCl3) δ : 5.62 - 6.50 (s, 2H), 5.34 (s, 1H), ; 13C NMR (126 MHz, CDCl3) The only carbon of the molecule is attached to boron and was not observed due to quadrupolar relaxation; 11B NMR (128 MHz, CDCl3) δ : 27.3.
26. A large magnetic stir bar is required as the reaction is heterogeneous.
27. Anhydrous magnesium sulfate was purchased from Fisher Chemical and used as received.
28. Pinacol was purchased from Oakwood Chemical and used as received.
29. Magnesium sulfate becomes a fine powder after the reaction is finished and filtration on a Büchner funnel is not efficient enough without a pad of Celite.
30. Granules for smooth boiling were purchased from Hengar Company.
31. An oil bath at 140 °C is used as source of heat.
32. Over the course of the distillation, crystallization may take place in the condenser to give a translucent solid.
33. A second reaction on the same scale (16.33 g of starting material) afforded 21.55 g of 2 (79%).
34. The product has been characterized as follow : bp = 108 °C at 23 mmHg; 1H NMR (600 MHz, CDCl3) δ : 5.35 (s, 1H), 1.33 (s, 12H); 13C NMR (151 MHz, CDCl3) δ : 24.6, 85.9, the carbon attached to boron was not observed due to quadrupolar relaxation; 11B NMR (193 MHz, CDCl3) δ : 28.4; FTIR (cm-1) (neat): 2982, 2935, 1470, 1408, 1360, 1274, 1213, 1169, 1137, 1111, 968, 901, 846, 823, 738, 672, 650, 577. HRMS (EI, Pos): calcd for C7H13BCl2O2 [M]+ : 210.0380 m/z, found 210.0383 m/z. Anal. calcd for C7H13BCl2O2: C, 39.87; H, 6.21. Found: C, 39.49; H, 5.95.
35. The product was determined to be 98.0% pure by quantitative NMR using ethylene carbonate as internal standard.
36. Sodium iodide was purchased from Oakwood Chemical and used as received.
37. Acetone (anhydrous) is purchased from Acros Organics in an AcroSeal bottle and used as received.
38. A Glindemann PTFE sealing ring is placed on the condenser joint to prevent any leak that could lead to evaporation of acetone.
39. The desired boronic ester 3 is sensitive to light due to its C-I bonds. For this reason work-up is done with the light of the hood switched off at all time. All manipulations outside the hood (including solvent evaporation and drying under a high-vacuum pump) are performed using aluminum foil around the flask.
40. Sodium thiosulfate pentahydrate was purchased from Anachemia and ground in a mortar before use.
41. Anhydrous magnesium sulfate is used to scavenge the water introduced from the sodium thiosulfate pentahydrate.
42. A large sintered funnel is necessary for the filtration of the large amount of solids added while quenching the reaction mixture.
43. A yellow oil is obtained that crystallizes to afford a pale yellow solid when the pressure is lowered to 20 mmHg. Crystallization can also be induced with a Pasteur pipette.
44. A second reaction on a similar scale (21.4 g of starting material) afforded 37.1 g of 3 (93%).
45. Initial filtration of the liquor was avoided due to continued crystal formation after filtration attempts. The liquor was instead gently removed via a Pasteur pipette. The cold hexanes that were used to wash the solid crystals were combined with this liquor and then evaporated to provide the solid for a second recrystallization.
46. The product has been characterized as follow: mp = 74 - 76 °C; 1H NMR (500 MHz, CDCl3) δ : 4.30 (s, 1H), 1.30 (s, 12H); 13C NMR (126 MHz, CDCl3) δ : 24.3, 85.5, the carbon attached to boron was not observed due to quadrupolar relaxation;11B NMR (128 MHz, CDCl3) δ : 29.8; FTIR (cm- 1) (neat): 2975, 2925, 2864, 1466, 1371, 1370, 1270, 1136, 1086, 963, 892, 842, 675, 661, 593, 575, 484; HRMS (EI, Pos): calcd for C 7H13BI2O2 [M]+: 393.9091 m/z, found 393.9105 m/z; Anal. calcd for C7H13BI2O2: C, 21.35; H, 3.33; Found: C, 21.40; H, 3.16.
47. A second recrystallization was performed on the solid obtained from evaporation of the filtrate (6.4 g) using 8 mL of hexanes, which was heated to 70 °C to induce dissolution. The desired boronic ester 3 (4.3 g) was isolated, giving an overall yield of 38.4 g (86%).
48. The product can be stored protected from light, under argon in the fridge for months, without degradation.




